Rice University researchers solve long-standing mystery about hemophilia protein
Rice University scientists have solved a long-standing mystery about where the body stores and deploys blood-clotting factor VIII, a protein that about 80 percent of hemophiliacs cannot produce due to genetic defects.
For years, conventional medical doctrine was that factor VIII was made in the liver, but studies over the past 10 years showed it was made in endothelial cells — the cells that line the walls of blood vessels — in the liver, heart, intestines and other organs. The new study, which is available online in the journal PLOS ONE, offers the first clear images of where factor VIII is stored within those cells. Researchers found the protein is both stored and secreted from a specialized organelle inside the cells that is also known to store and deploy another important blood-clotting protein called “von Willebrand factor” or VWF.
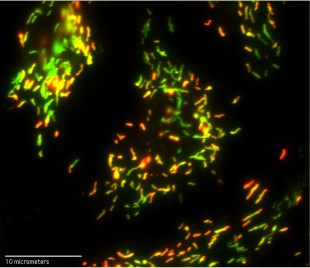
At 100X magnification, this optical microscope image reveals fluorescently tagged antibodies that are bound to clotting factor VIII (red) and von Willebrand factor (VWF) (green) inside organelles known as Weibel-Palade bodies in human umbilical vein endothelial cells. Yellow areas contain roughly equal amounts of factor VIII and VWF. Credit: N. Turner/Rice University
Hemophilia A, a bleeding disorder that affects almost one in 5,000 men, is caused by genetic defects that eliminate or reduce a person’s ability to make factor VIII. The protein is one of several that are required for effective blood clotting; without it, patients can suffer prolonged bleeding and death even from minor cuts.
“Great clinical advances have been made over the past 50 years in spite of our lack of understanding of where factor VIII was made and stored,” said study co-author Dr. Joel Moake, a hematologist with appointments in Rice’s Department of Bioengineering and Baylor College of Medicine in the Texas Medical Center. “Understanding how the body makes, stores and deploys the protein will be increasingly important in the future as physicians look to develop gene therapies that could free patients from a reliance on regular injections of factor VIII.”
Hemophilia is caused by recessive genetic defects on the X chromosome, which means that men typically suffer from hemophilia, and women usually act as carriers. Hemophilia A, the more common form of the disease, accounts for about 80 percent of known cases. Hemophilia B, the less common form, is caused by defects in clotting factor IX; the disorder is widely known to have affected Queen Victoria’s descendants.
Factor VIII is a prominent component of a variety of specialized proteins involved in clot regulation. Some of these signal where wounds occur, others attract clot-initiating cells called platelets and many act only to break up clots or destroy their clot-forming relatives. Factor VIII, a signaling protein, is one of several that act in concert to produce a signaling “cascade,” an amplification process that allows the body to quickly transform a weak signal from a tiny cut into a blaring clarion call to rapid action.
Factor VIII was identified in the 1950s, and clinicians have used it to treat patients for decades, both by isolating it from donated blood and by producing it through biotechnology. Today, many hemophiliacs live symptom-free, thanks to regular injections of factor VIII.
The factor VIII research in Moake’s lab at Rice’s BioScience Research Collaborative began in early 2014 based on a hunch by lead author Nancy Turner. Turner, a research biochemist, specializes in the study of endothelial cells. Though all endothelial cells are similar, the human body makes more than a dozen varieties. Each organ has its own special types of endothelial cells, and Turner has become intimately familiar with several of them over the past 25 years.

Rice bioengineering researchers Nancy Turner and Joel Moake discovered that blood-clotting factor VIII is both stored and secreted from endothelial cells, which make up the inner lining of blood vessels.
“Endothelial cells are the gate controllers for the blood system, and to me, they make all the exciting proteins,” she said. “They’re right on the surface next to the blood, and they constantly interact with the plasma. Similar to circulating cells, they produce their own defensive proteins to fight infections.”
Though Turner had not previously studied factor VIII, she had done extensive experiments on VWF, which is made in endothelial cells and stored in specialized organelles called “Weibel-Palade bodies.” VWF and factor VIII are often bound together.
Based on her prior work, Turner was intrigued by a pair of 2014 studies that examined factor VIII in mouse endothelial cells and found that factor VIII was produced in the endothelial cells within the liver, and not in the liver cells.
“They stopped short of saying that factor VIII was stored in endothelial cells, but they suggested the possibility,” she said. “One of the papers was very elegant, and I liked it a lot, but it made a statement that really bothered me.”
The study explained that factor VIII had been found in a half-dozen types of endothelial cell types, but never in “human umbilical vein endothelial cells,” or HUVECs (pronounced: HUE-vecks).
“HUVECs are the generic human endothelial cells that (biological researchers) use the first time they do anything,” Turner said. “They’re cheap. They’re easy to work with, and they’ve been the model for endothelial cells for, I don’t know, at least 50 years.
“So far, everything I’ve ever looked at in endothelial cells has been consistent. The different types might have different amounts of something, but they’re very similar. There hasn’t been anything that was wholly different from one type to another. Not yet.
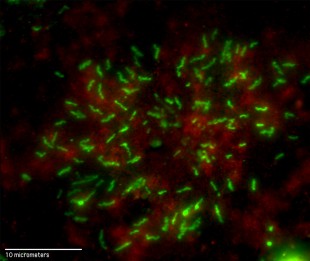
This 100X magnification from an optical microscope shows fluorescently tagged antibodies that are bound to clotting factor VIII (green) in human umbilical vein endothelial cells. Red areas show a cellular structural protein called beta-actin. Credit: N. Turner/Rice University
“So I thought, ‘OK, fine. I’m already doing gene-expression experiments, I might as well just throw factor VIII in there and see.”
The experiments she was conducting involved both HUVECs, the type of endothelial cell found in large veins, and “glomerular microvascular endothelial cells,” or GMVECs (pronounced: JIM-vecks), which are found in the smallest capillaries of the kidney. Turner was conducting an extensive analysis to see how protein production differed in the two varieties.
She told Moake she wanted to include the factor VIII gene in their current study, and he was both cautious and encouraging because many other researchers had tried for decades and had not found factor VIII in HUVECs. He also believed that Turner was clever enough to succeed where others had failed.
Turner examined the cells for factor VIII messenger RNA and found that it was present, which meant it was possible that the cells were making the protein. She next ordered a specific antibody that was designed to detect factor VIII. The antibody contained a fluorescent dye that would show up clearly under a microscope if the antibody detected any factor VIII.
“I did the experiment and looked, and not only was it there, but it was bright, easy to see, and it worked perfectly the first time,” Turner said. “I thought, ‘What have people been doing for 20 or 30 years? Why couldn’t they see this?’ And then I thought, ‘This was too easy. No one is going to believe me.'”
In fact, the early success was so unexpected that Turner at first doubted herself and immediately set about running controls to rule out mistakes. Were the antibodies interfering with one another? Was there any contamination? Was the microscope working correctly? Could the reading be a false positive, an inadvertent result of another reaction she hadn’t expected?
“No. 1, I had to convince myself,” she said. “I am always skeptical if something is too easy. I have reviewed many, many papers, and I can always find what people do wrong.”
Once she had convinced herself, she and Moake had to convince the paper’s referees. As she’d expected, they were skeptical. She said the bulk of the work over the past year involved doing a number of controls to remove any doubts about the findings. In the end, the research confirmed that factor VIII was made in both HUVECs and GMVECs. Moreover, Turner found that factor VIII, like VWF, is both stored and secreted from Weibel-Palade bodies.
Moake said the discovery has clear implications for any future treatments that aim to repair the genetic defects in patients with hemophilia A.
“Now that we recognize that factor VIII is normally synthesized in endothelial cells and stored in Weibel-Palade bodies, those become the precise, most effective physiological targets for gene delivery,” Moake said.
The research was supported by the Mary Rodes Gibson Foundation, the Hinkson Memorial Fund and Rice University.


Leave a Reply